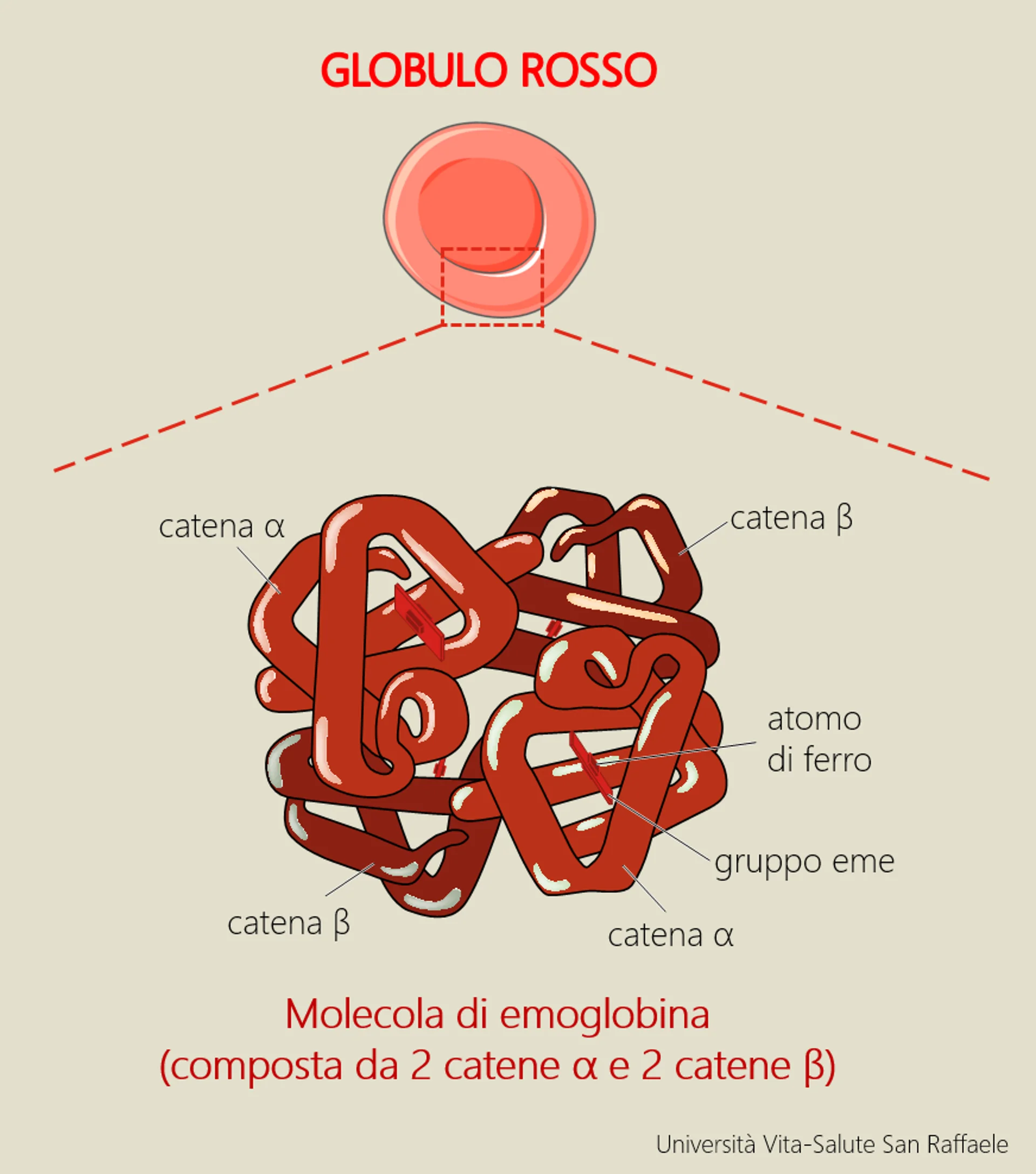
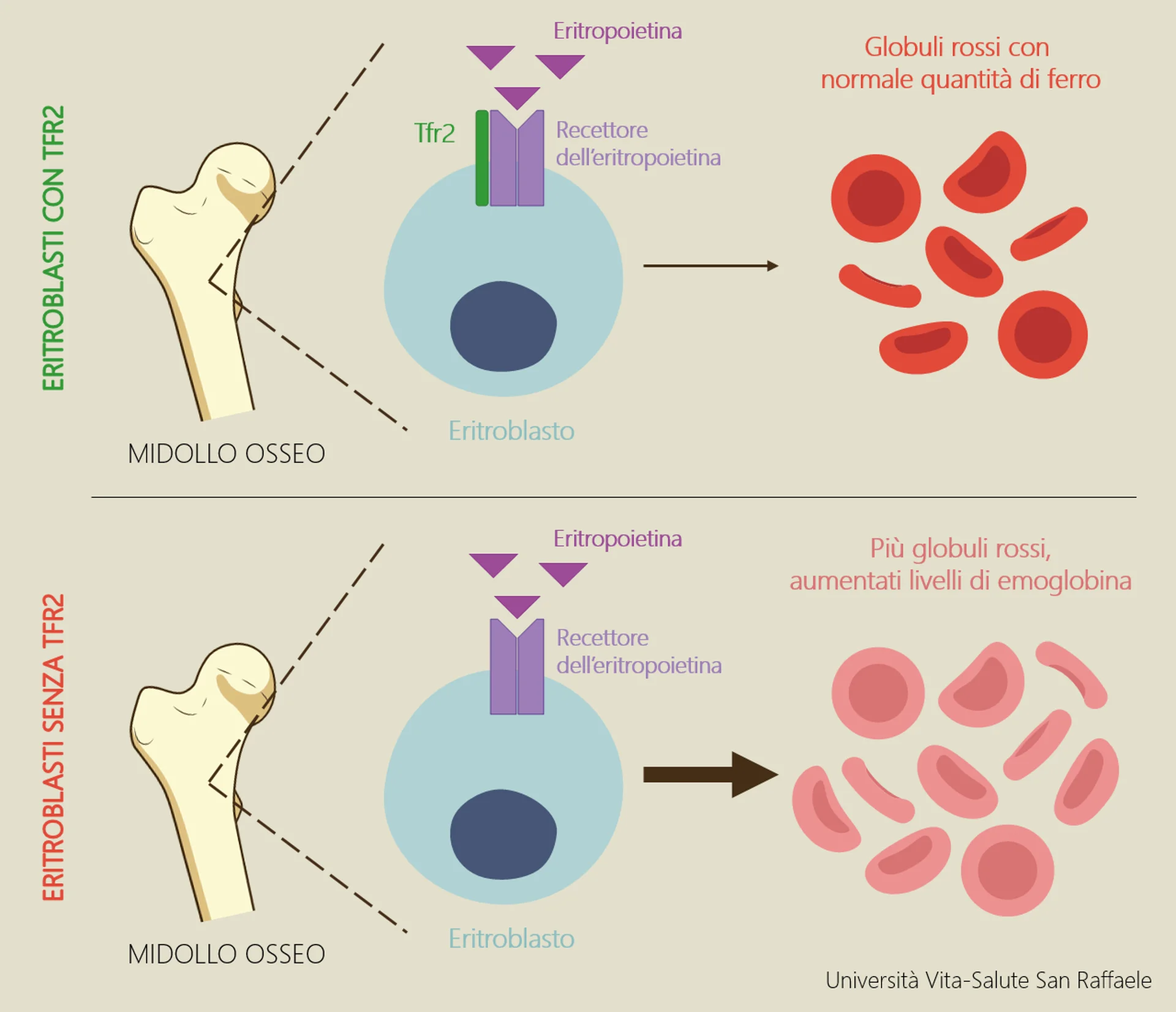
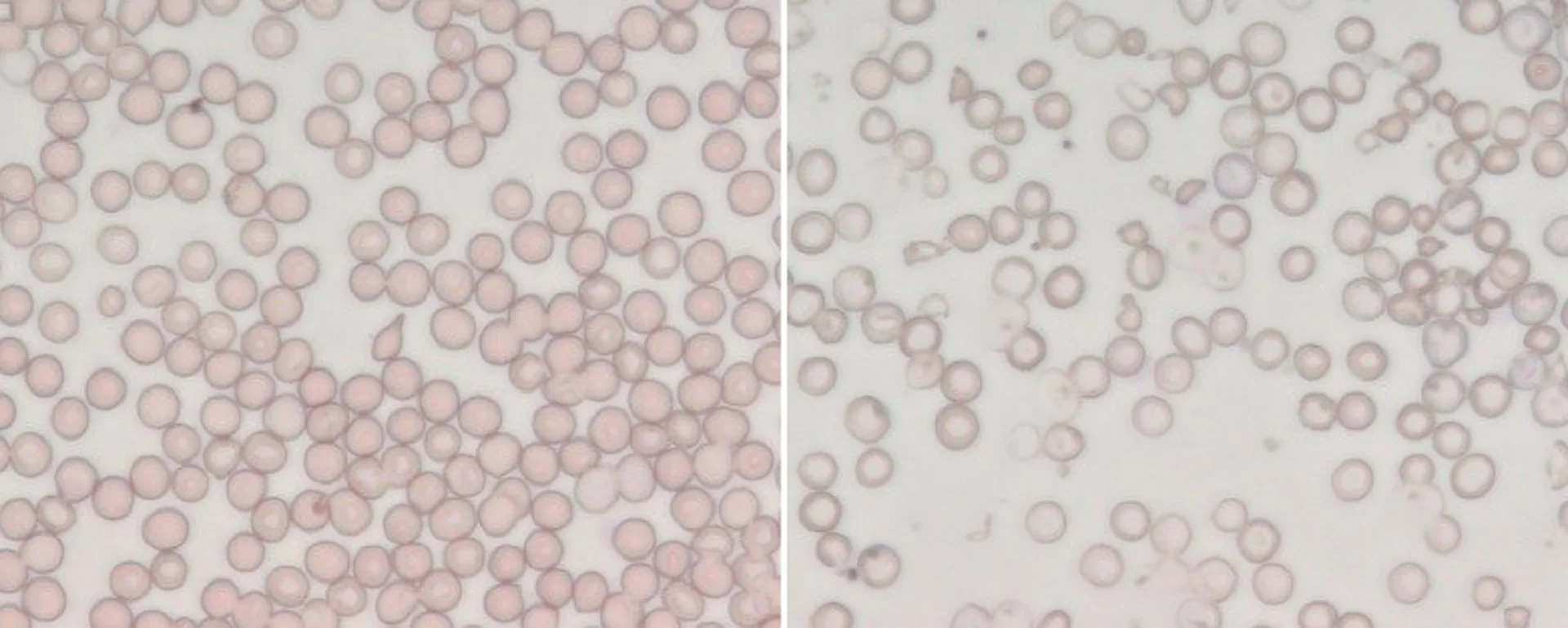
Therefore, the aim of the project is to investigate and unveil the molecular role of Tfr2 on ineffective erythropoiesis in beta-thalassemia. Preliminary data suggest that in a model of thalassemia intermedia (not dependent on blood transfusions) the absence of Tfr2 leads to a significant improvement of anemia. “The ultimate goal is to identify and test a molecule that selectively inhibits Tfr2 in thalassemic animals to be proposed as a novel therapeutic agent to control ineffective erythropoiesis and improve beta-thalassemia anemia”.
The researchers note that, in the long term, the beneficial effect of the increased erythropoiesis could be limited, probably because of the consumption of iron, which becomes insufficient for the enhanced erythropoiesis. “For this reason, during the project we will pay particular attention to the modulation of iron levels, a critical aspect of this disorder”.
Finally, considering that the preliminary studies have been carried out in the thalassemia intermedia model, “we would like to validate the same approach also in thalassemia major, the most severe form of the disease”.