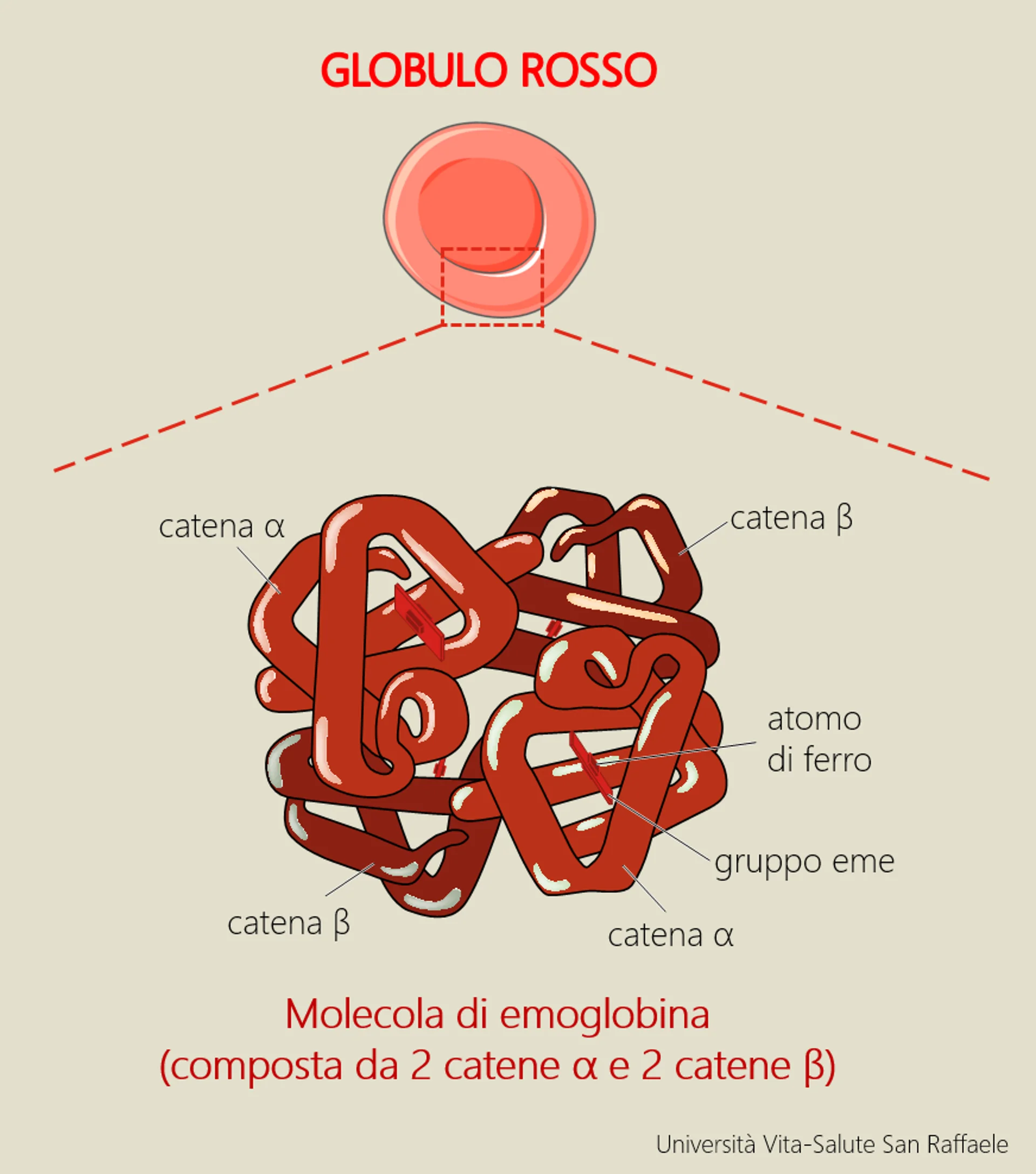
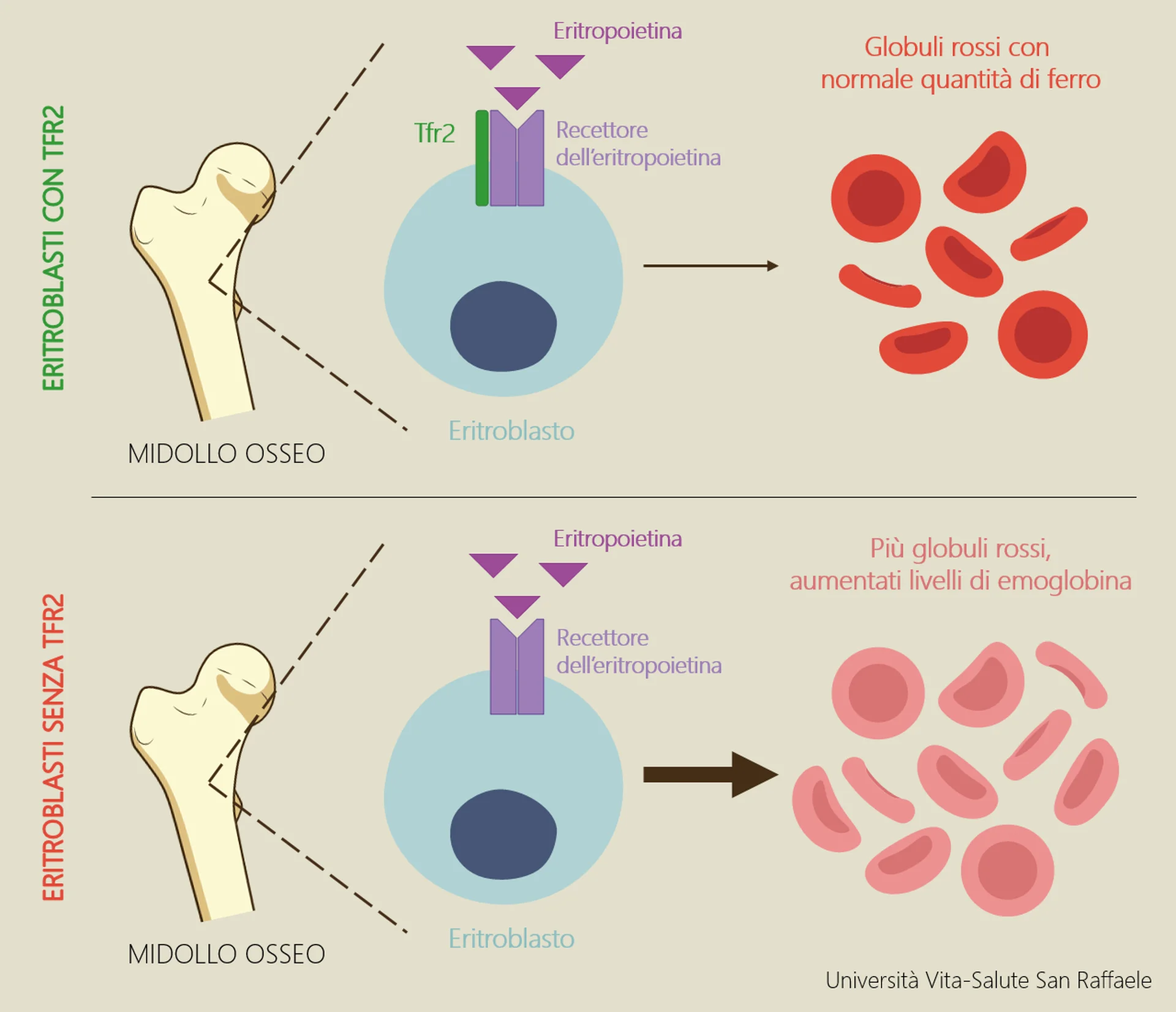
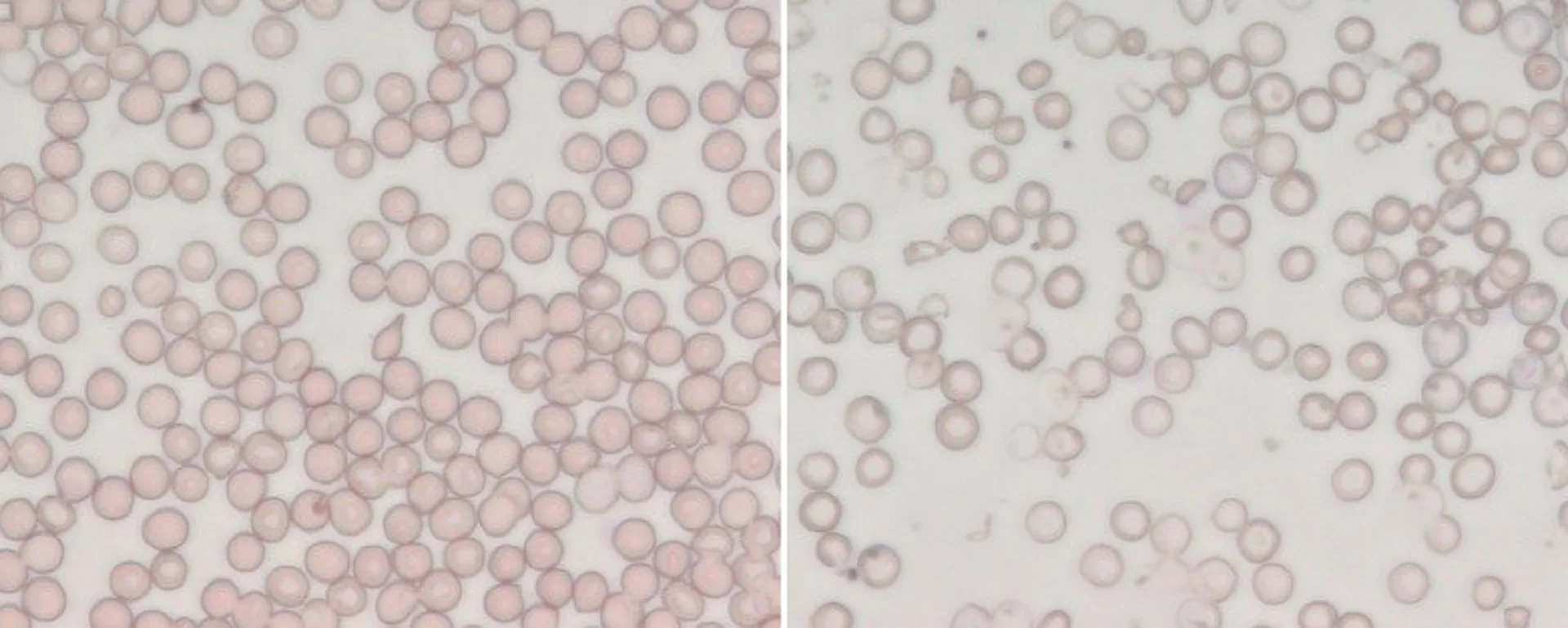
La beta-talassemia (o più correttamente al plurale, le beta-talassemie) sono disordini causati da mutazioni nel gene della beta-globina; le due forme principali sono la talassemia minor (causata da una mutazione nel gene della beta-globina, con conseguente modesta riduzione della sintesi della catena β) e la talassemia major (quando si verificano invece due mutazioni nel gene della beta-globina, con grave riduzione o assenza totale della catena β). Esiste inoltre una forma definita talassemia intermedia, la cui severità è appunto intermedia tra le forme sopra descritte. Spiega Antonella: “In mancanza di quantità adeguate di catena β, nei globuli rossi si formano depositi di catena α: ne derivano globuli rossi deformati, anemia, eritropoiesi (il processo di formazione dei globuli rossi) anomala, sovraccarico di ferro, e nei casi più gravi anche splenomegalia (ingrossamento della milza) e deformità delle ossa”.
Le terapie ad oggi utilizzate si basano soprattutto sul sottoporre i pazienti a continue trasfusioni di sangue associate a trattamenti ferrochelanti, che consistono nell’assunzione di farmaci che legano ed eliminano il ferro in eccesso o nel trapianto di midollo osseo quando un donatore è disponibile. Nuove terapie per la talassemia sono in fase sperimentale. Il gruppo della Prof.ssa Camaschella, per anni Ordinario di Medicina Interna presso l’Università Vita-Salute San Raffaele, e in particolare la Dott.ssa Nai collaborano da molto tempo con la Prof.ssa Giuliana Ferrari, Ordinario di Biologia Molecolare presso l’Università Vita-Salute San Raffaele e Capo Unità dell’Istituto San Raffaele Telethon per la Terapia genica (SR-TIGET), nel cui laboratorio la principale linea di ricerca è rappresentata dalla terapia genica della talassemia. “La sperimentazione clinica in pazienti affetti da talassemia major trasfusione-dipendente, sta dando dei risultati incoraggianti, in special modo nei pazienti più giovani, ma si tratta di un approccio complesso e costoso, dunque c’è molto interesse nell’identificazione di strategie alternative, di più semplice impiego che possano essere più efficaci a largo spettro” specifica la Dr.ssa Nai.