La pandemia in corso di COVID-19 ha imposto alla comunità scientifica di dedicarsi alla ricerca e sviluppo di varie strategie per combattere l’infezione da SARS-CoV-2. L’unico modo informato per identificare nuovi antivirali passa attraverso la conoscenza della struttura tridimensionale, letteralmente la forma, delle proteine del virus che sono responsabili dell’infezione delle cellule e della replicazione del virus. Una volta nota la struttura delle proteine, è possibile per i chimici sintetizzare dei composti che ne bloccano la funzione, e di conseguenza il ciclo vitale del virus.
Ne parliamo in questo ciclo di 5 puntate con l’aiuto del Dott. Massimo Degano, Group leader dell’Unità di Biocristallografia dell’IRCCS Ospedale San Raffaele e docente presso il nostro Ateneo (insegna Chimica al Corso di Laurea in Odontoiatria e Protesi dentaria e all’International MD Program, Biochimica al Corso di Laurea in Medicina, e Biologia strutturale al Corso di Laurea in Ricerca Biotecnologica in Medicina).

Abbiamo spiegato nelle puntate precedenti che per sviluppare farmaci contro SARS-CoV-2 è necessario conoscere con elevato dettaglio (risoluzione) la struttura delle proteine del virus che abbiamo scelto come bersaglio sulla base della loro importanza per il ciclo vitale del virus. Questo è il compito della biologia strutturale, e abbiamo già spiegato il principio di funzionamento della criomicroscopia elettronica. La prima tecnica che ha consentito di visualizzare con dettaglio atomico le strutture delle molecole, e tuttora fornisce un dettaglio elevatissimo, è la cristallografia a raggi X.
I successi (e i contributori) della cristallografia a raggi X
La teoria della cristallografia è stata sviluppata poco tempo dopo la scoperta dei raggi X da parte di Wilhelm Röntgen, grazie ai contributi di Max von Laue (premio Nobel nel 1914) e William Henry e William Lawrence Bragg (padre e figlio, entrambi insigniti del premio Nobel nel 1915), e negli anni è stata costantemente affinata e migliorata. Se al tempo di questi pionieri determinare la struttura di piccole molecole come l’aspirina era una sfida formidabile, grazie a visionari quali furono sir John Kendrew e Max Perutz si è riusciti a determinare le prime strutture di proteine intere, mioglobina ed emoglobina. Dal 1945 ad oggi la lista dei successi della cristallografia nel fornirci le immagini di proteine di interesse biomedico è infinita, e l’importanza di queste informazioni sono corroborate dall’Accademia reale svedese delle scienze che regolarmente premia con il Nobel gli studi strutturali che ci consentono di capire il funzionamento di proteine complesse (https://www.iucr.org/people/nobel-prize).
Usare i raggi X per visualizzare la struttura delle grandi molecole biologiche
Quali sono i passaggi di questa tecnica? Osserviamo lo schema:
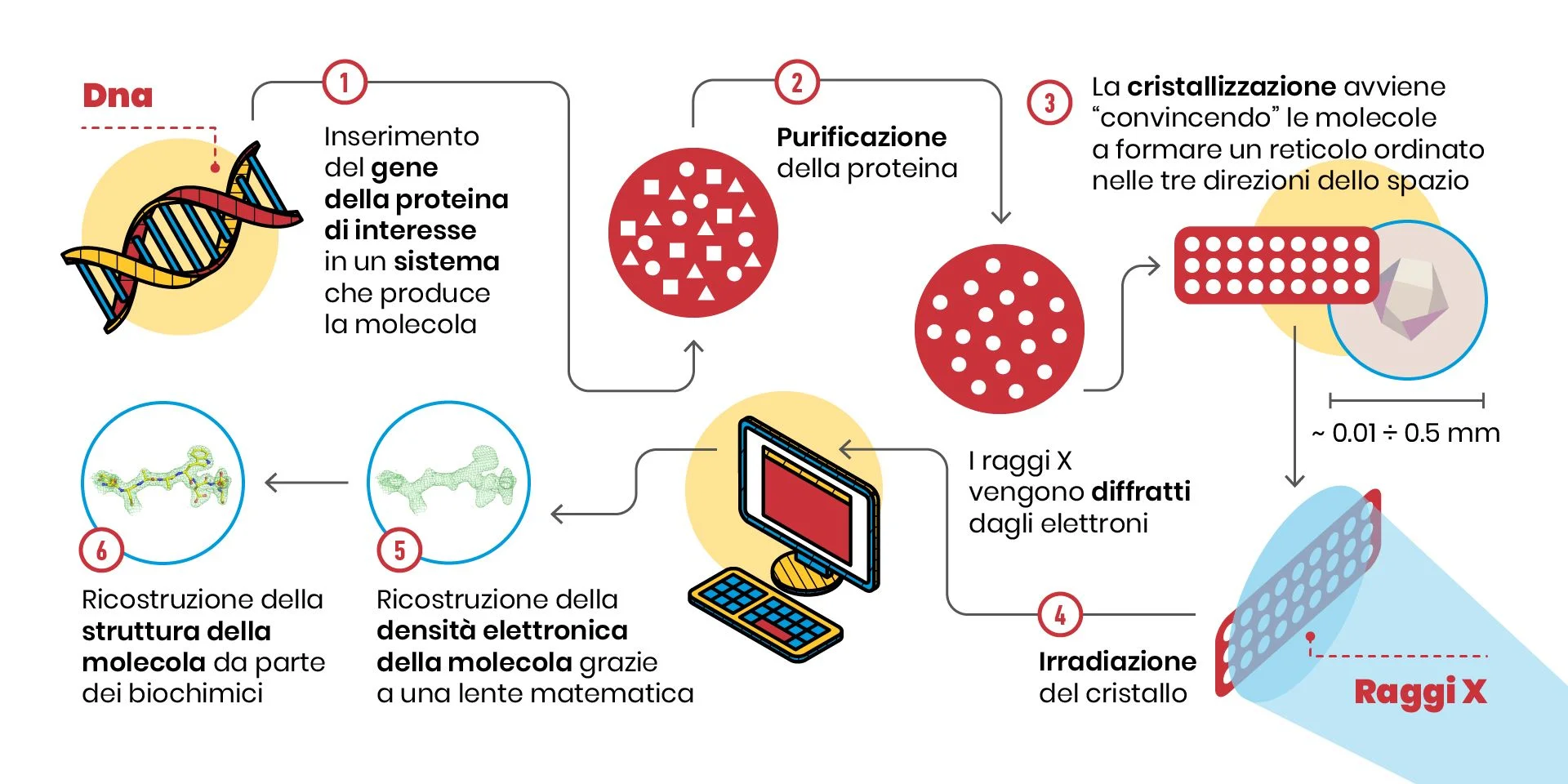
1, 2. Prima di tutto occorre avere a disposizione milligrammi di campione puro, cioè quantità infinitesime di contaminanti: in pratica, tantissime molecole tutte uguali.
3. Poi, occorre far sì che queste molecole formino un cristallo, cioè un reticolo tridimensionale ordinato. Questo passaggio è fondamentale, perché le molecole nel cristallo sono posizionate secondo schemi simmetrici, come quelli studiati da M. C. Escher (https://mcescher.com/gallery/symmetry/).
4. L’ordine delle molecole nel cristallo fa sì che gli elettroni attorno agli atomi, quando irradiati con i raggi X, diffrangano la radiazione in fase, sommandosi come le componenti di un coro di voci. Questo effetto di amplificazione ci consente di misurare lo spettro di diffrazione, ovverosia che direzione abbiano e quanto siano intensi siano i raggi X diffratti dalle molecole nel cristallo.
5. Una volta fatta questa misura, e applicando una sorta di lente matematica (chiamata “la trasformata inversa di Fourier”), si ottiene la mappa di densità elettronica che ci dice come siano disposti gli atomi nel cristallo. La densità elettronica è visualizzabile al computer, e in essa si devono inserire le componenti della molecola biologica che stiamo studiando (amminoacidi per le proteine, ribosio e basi azotate per DNA e RNA…), in un processo ad incastro che a molti potrebbe ricordare qualche videogioco.
6. Completata l’interpretazione della densità elettronica, abbiamo finalmente la struttura tridimensionale!
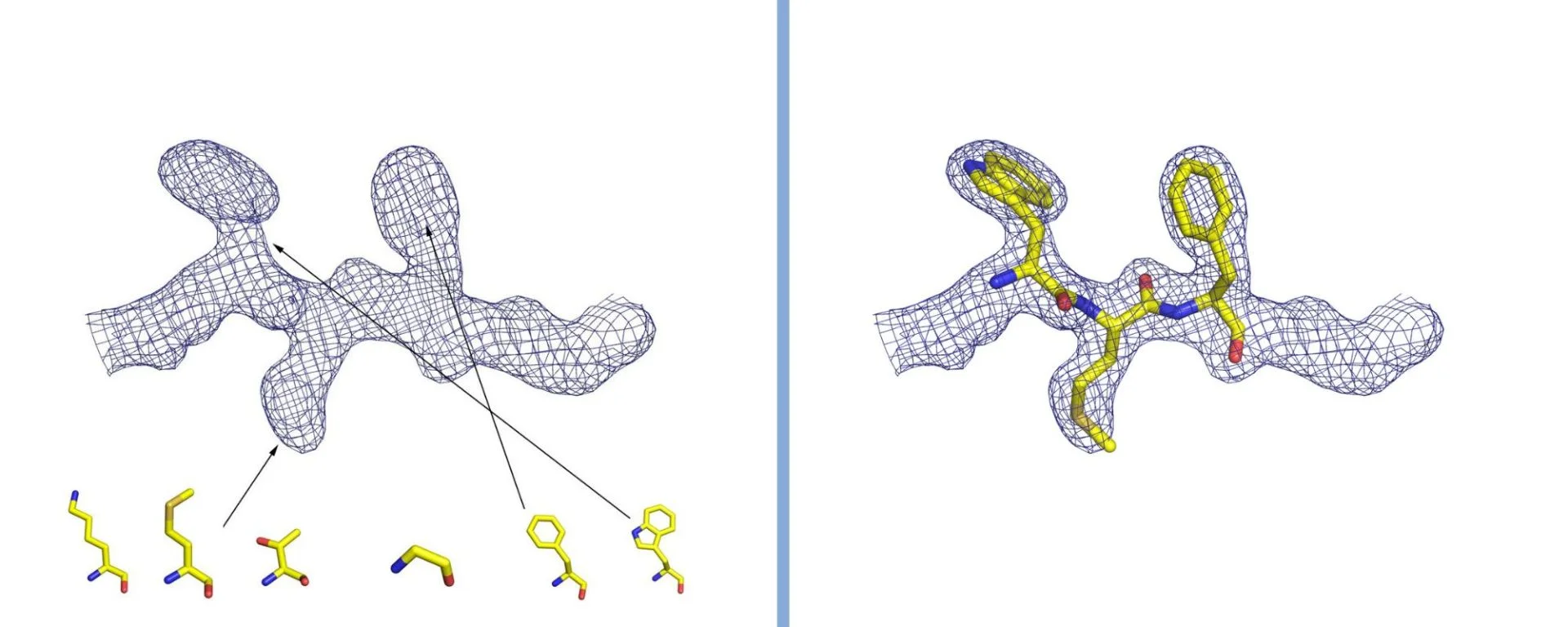
Interpretazione della densità elettronica in cristallografia. La densità è rappresentata con una griglia, al cui interno si trovano gli atomi che compongono la proteina. Nel caso di una proteina, si osserva la forma della densità e la si confronta con la struttura dei venti amminoacidi che la costituiscono (alcuni dei quali sono rappresentati in figura). Inserendo gli amminoacidi nell’ordine corretto e seguendo l’andamento nelle tre dimensioni della densità elettronica si ricostruisce la struttura della proteina. Gentile concessione del Dott. Degano.
Utilizzi della cristallografia
La cristallografia è molto versatile ed è uno strumento potentissimo per lo sviluppo di composti guida con prospettive di divenire farmaci. Con varie metodologie è possibile ottenere la struttura delle proteine bersaglio legate a molecole con attività inibitoria: si può infatti “impregnare” i cristalli in una soluzione in cui è disciolta una sostanza che sappiamo può legarsi alla proteina bersaglio, oppure ottenere cristalli della proteina dopo averla legata al composto. Di questo abbiamo visto un esempio quando abbiamo parlato della proteasi principale Mpro di SARS-CoV-2. Una volta nota la struttura della proteina legata a un composto, possiamo visualizzare quali parti della proteina (che possiamo assimilare ad un guanto) non vengono correttamente riempiti dalla molecola (le dita di una mano). Con questa informazione possiamo modificare la molecola inibitore per far sì che complementi meglio possibile il nostro bersaglio, e ne blocchi la funzione!
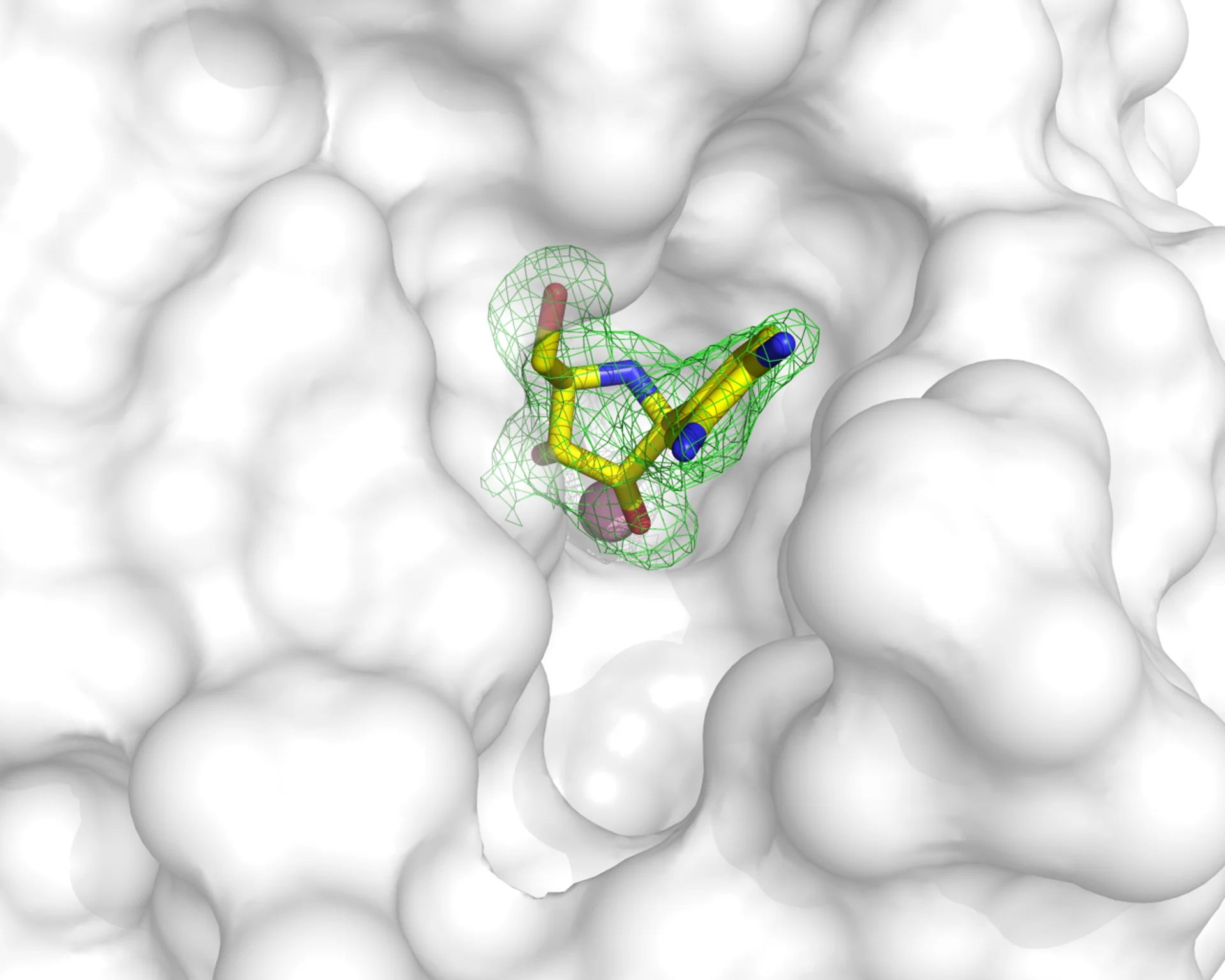
Esempio di inibitore di un enzima che è stato inserito nelle molecole di un cristallo. La densità elettronica in verde definisce chiaramente dove si posiziona la molecola nella struttura proteica, rappresentata con la superficie bianca. Si può notare come la molecola non occupi perfettamente la cavità disponibile, per cui si può ingegnerizzare un composto che complementi meglio la struttura proteica e ne blocchi efficientemente la funzione. La struttura utilizzata è accessibile con il codice 3MKN nel Protein Data Bank. Gentile concessione del Dott. Degano.
A settimana prossima, con la nostra quinta e ultima puntata sulla struttura delle proteine!
Leggi gli altri episodi de "La biologia strutturale per SARS-CoV-2"
Prima puntata: “La proteina Spike”
Terza puntata: “Sviluppare antivirali in modo razionale: “spuntare” le forbici di SARS-CoV-2”
Quinta puntata: “RNA polimerasi, la “fotocopiatrice distratta” di SARS-CoV-2”
Ti potrebbero anche interessare

Malattia IgG4-correlata: lo studio su obexelimab coordinato da UniSR

Carcinoma renale, l'intelligenza artificiale stima il rischio pre-chirurgia

Trapianto di midollo osseo e leucemia: conoscere i meccanismi delle recidive aiuta a prevenirle